Computational Chemistry¶

abinit
abinit is a DFT code based on pseudopotentials and a planewave basis, which calculates the total energy, charge density and electronic structure for molecules and periodic solids. In addition to many other features, it provides the time dependent DFT, or many-body perturbation theory (GW approximation) to compute the excited states.




cp2k
cp2k is a quantum chemistry and solid state physics software package that can perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. CP2K provides a general framework for different modeling methods such as DFT using the mixed Gaussian and plane waves approaches GPW and GAPW.

dalton
dalton is a general-purpose program system for advanced quantum-chemical molecular electronic-structure calculations. With Dalton, molecular systems may be studied using a variety of electronic-structure methods, including the Hartree–Fock (HF) and Kohn–Sham (KS) self-consistent-field (SCF) methods for wide applicability, the multiconfigurational SCF (MCSCF) method for high flexibility, and various coupled-cluster (CC) methods for high accuracy.



elk
elk is an all-electron full-potential linearised augmented plane-wave (LAPW) code for determining the properties of crystalline solids. It is extensively used for materials which are particularly sensitive to the types of approximation used or for which pseudopotential methods are not appropriate.


flower
FlowER uses flow matching to model chemical reaction as a process of electron redistribution, conceptually aligns with arrow-pushing formalisms. It aims to capture the probabilistic nature of reactions with mass conservation where multiple outcomes are reached through branching mechanistic networks evolving in time.


gaussian
gaussian 16 is the latest in the Gaussian series of programs. It provides state-of-the-art capabilities for electronic structure modeling. Gaussian 16 is licensed for a wide variety of computer systems. All versions of Gaussian 16 contain every scientific/modeling feature, and none imposes any artificial limitations on calculations other than your computing resources and patience.


nwchem
nwchem aims to provide its users with computational chemistry tools that are scalable both in their ability to treat large scientific computational chemistry problems efficiently, and in their use of available parallel computing resources from high-performance parallel supercomputers to conventional workstation clusters.

octopus
octopus is a scientific program allowing to describe non-equilibrium phenomena in molecular complexes, low dimensional materials, and extended systems. In usual applications, electrons are described quantum-mechanically within density-functional theory (DFT), or in its time-dependent form (TDDFT) when doing simulations in time, using a real-space grid, while nuclei are described classically as point particles. Electromagnetic fields can be treated either classically or quantum mechanically within a generalized time-dependent density functional theory.

openbabel
openbabel is a free, open-source version of the Babel chemistry file translation program. Open Babel is a project designed to pick up where Babel left off, as a cross-platform program and library designed to interconvert between many file formats used in molecular modeling, computational chemistry, and many related areas.

orca
orca is an ab initio quantum chemistry program package that contains modern electronic structure methods including density functional theory, many-body perturbation, coupled cluster, multireference methods, and semi-empirical quantum chemistry methods. Its main field of application is larger molecules, transition metal complexes, and their spectroscopic properties.
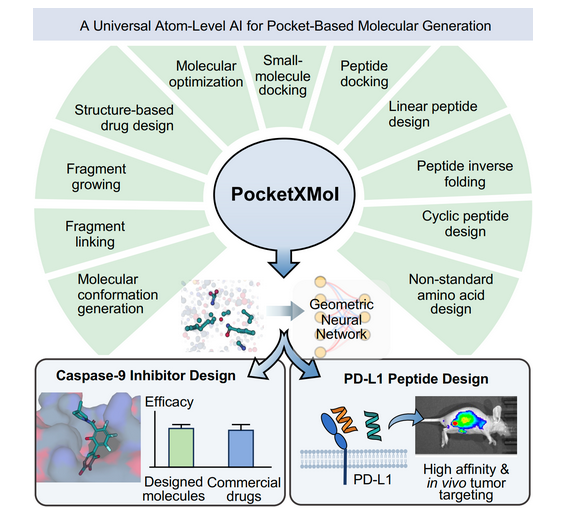
pocketxmol
pocketxmol is an AI generative model that learns fundamental atom interactions, enabling applications governed by atom interactions within a pocket, including, small-molecule docking, peptide docking, and molecular conformation generation, structure-based drug design (SBDD), fragment linking/growing, PROTAC design, de novo linear/cyclic peptide design, and peptide inverse folding.

psi4
psi4 provides a wide variety of quantum chemical methods using state-of-the-art numerical methods and algorithms. Several parts of the code feature shared-memory parallelization to run efficiently on multi-core machines (see Sec. Threading). An advanced parser written in Python allows the user input to have a very simple style for routine computations, but it can also automate very complex tasks with ease.

quantum espresso
quantum espresso (qe) is an integrated suite of Open-Source computer codes for electronic-structure calculations and materials modelling at the nanoscale. It is based on density-functional theory, plane waves, and pseudopotentials.

